Types of SMA
Genetics
DIAGNOSIS
TREATMENT AND CURE
Types of SMA
The severity of SMA varies greatly between individuals, so the disease is divided into four types. These are referred to as SMA type I, SMA type II, and SMA type III and SMA type IV. The type of SMA is determined based on the age at which symptoms appear, the related severity of muscle weakness, and the highest motor function a child achieves.
Even within these types there is a wide spectrum of severity as SMA affects everyone differently.
SMA Type I (Werdnig–Hoffmann disease)
It is the most severe form of SMA and defined as children who are not able to sit independently. It is usually evident at birth, or in the first few months afterwards (0-6 months). Symptoms include floppy limbs and weak trunk movement. They will also have a hard time feeding and swallowing, holding their head up, and breathing. They may need breathing assistance or a feeding tube. Type 1 SMA progresses rapidly, with the weakening of muscles leading to frequent respiratory infections and usually death by the age of 2.
SMA Type II(Dubowitz disease)
Symptoms of SMA type 2 appear between seven months and 18 months of age. Children are diagnosed with SMA type 2 if they were strong enough to maintain sitting position at some point, and some may have achieved the ability to stand, but they never gain the ability to walk unaided. The rate of progression can vary greatly. Just like in SMA type 1, children and adults with SMA type 2 often have difficulties swallowing and are prone to respiratory infections due to weakness of the muscles used for breathing. They also require mobility aids (for example, a wheelchair) due to progressive muscle weakness. However, with good care, they live to become teenagers and adults. Stronger people with SMA 2 may work, and some start families.

SMA Type III (Kugelberg–Welander disease)
Symptoms for this type can first appear from 18 months to early adulthood. Patients with Type 3 SMA can stand and walk but may have trouble getting up from sitting position, running and climbing stairs. They may also experience mild muscle weakness and are at greater risk for respiratory infections. Later in life, they may need a wheelchair to get around. Most patients with Type 3 SMA have a life expectancy close to normal.

SMA Type IV
SMA type 4 is rare. Symptoms for this rare type of SMA do not usually emerge until the second or third decade of life. Patients with Type 4 SMA can walk during adulthood but will usually experience slowly progressive muscle weakness and other typical SMA symptoms.
Overview of SMA clinical classification
| SMA Type | Usual age of symptoms | Impact of muscle weakness on sitting / walking |
| Type 1 | Younger than 6 months | Unable to sit or roll independently |
| Type 2 | 6 – 18 months | Able to sit but not walk independently |
| Type 3 | 18 months – 3 years | Able to walk, though may lose this ability over time. |
| Type 4 | Over 18 years | Mild walking difficulties |
Adapted from Tillmann et al. 2018
Genetics
SMA is a genetic neuromuscular disease that is passed down through families.Genetic conditions are caused by differences or ‘faults’ in our genes.
Genes involved in SMA:
The SMN1 gene: SMA is caused by a mutation(an unusual change or ‘fault’ in the genes is known as a mutation) in the Survival Motor Neuron Gene 1 (SMN1). In a healthy person, two copies of the SMN1 gene are present and it produces a protein—called survival motor neuron protein or SMN protein—that is critical to the function of the nerves that control our muscles. Without it, those nerve cells cannot properly function and eventually die, leading to debilitating and often fatal muscle weakness.
People with SMA have two faulty copies of the SMN1 gene. This means they are unable to produce enough SMN protein to have healthy lower motor neurons.
The SMN2 gene: A second gene also has a role in producing SMN protein. This is the Survival Motor Neuron 2 (SMN2) gene, sometimes referred to as the SMA “back-up gene”.
SMN2 has an important single base (nucleotide) difference from SMN1. This causes a small chunk of the gene, called Exon 7, to be excluded in the majority of SMN protein that the SMN2 gene makes. It is estimated that only about 10% of the SMN protein made from SMN2 is functional.
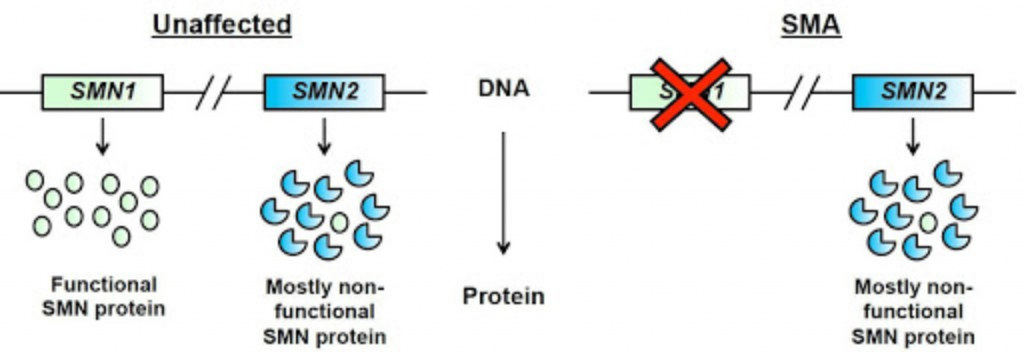
Figure adapted from Burghes, A.H. and Beattie, C.E.
Unlike most genes, the number of copies of SMN2 on each chromosome can vary from one person to the next.
As the severity of a person’s SMA has been linked to how much SMN protein a person makes, there is therefore a broad relationship between the number of SMN2 copies a person has (“SMN2 copy number”) and the likely severity of their symptoms. Having more SMN2 copies is generally associated with less severe SMA symptoms. However, accurate predictions cannot be made about the Type or severity of SMA based on the SMN2 copy number alone. This is likely to be because other genetic factors also have a modifying influence.
Inheritance of these Genes
People have 23 pairs of chromosomes. SMA is an autosomal recessive condition. SMA, the Survival Motor Neuron 1 (SMN1) gene is located on the fifth autosomal chromosome, in the region labelled ‘q’. Almost all patients (>95%) with the most common forms of SMA have deletions of SMN1 exon 7; the loss of exon 7 abolishes production of SMN1 protein.
Autosomal conditions affect both males and females equally.
In an autosomal recessive condition like SMA:
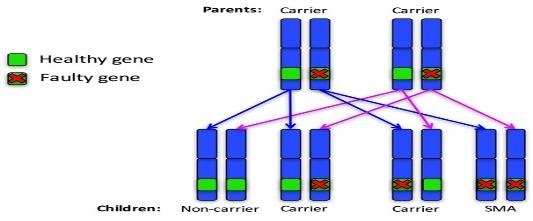
a person will only have SMA if they inherit two faulty copies of the SMN1 gene.
A person who carries one faulty copy of the gene and one healthy copy will not have the condition but is a carrier. They do not have any symptoms, but the faulty gene can be passed on to their children.
How will this affect the children?
The chances of the children being carriers or having SMA will depend on whether you or your partner have SMA or are carriers. The chances stay the same for each pregnancy; having one child who has SMA or is a carrier doesn’t change the chances for any further children. Each copy of the gene (healthy or faulty) has the same chance of being passed on. This happens randomly, like the result of a coin toss.
The following diagrams show what the chances are in different families.
For the purpose of the diagrams, a ‘non-carrier’ means a person who does not carry the faulty gene and does not have SMA.
Autosomal recessive family 1: Both parents are carriers
For each pregnancy, the chances are:
- Child will not have SMA and won’t be a carrier: 1 in 4 chance (25%)
- Child will not have SMA but will be a carrier: 2 in 4 chance (50%)
- Child will have SMA: 1 in 4 chance (25%)
Autosomal recessive family 2: One parent is a carrier the other is a non-carrier
For each pregnancy, the chances are:
- Child will have SMA: not possible
- Child will not have SMA and won’t be a carrier: 2 in 4 chance (50%)
- Child will not have SMA but will be a carrier: 2 in 4 chance (50%)
DIAGNOSIS
SMA is usually diagnosed in one of three ways:
- Through genetic testing, after an infant or child shows symptoms of SMA
- Through a positive newborn screening result
- Through prenatal testing
Early Symptoms of SMA
A doctor may suspect SMA when a child is noticeably weak or has a delay in meeting developmental milestones. This may include delays in holding their head up, rolling over, sitting independently, standing, or walking.
SMA is not the only condition that can cause weakness or a delay in meeting milestones, so further examination and testing is needed to make an SMA diagnosis.
If a doctor suspects SMA, they may:
Order genetic testing through a blood sample, or
Refer the child to a neurologist who will also perform an examination, and then order genetic testing (again through a blood sample) to confirm the diagnosis.
A simple blood draw test can identify an estimated 95% of all SMA cases. The other 5% are caused by a rare mutation and must be identified through further testing.
Newborn Screening
When a baby is born, a small blood sample is taken. This sample is then screened for a number of genetic conditions. In 2018, some states began screening infants for SMA. If a newborn screening result is positive for SMA, follow-up testing is required to confirm the diagnosis.
Newborn screening allows an infant to begin treatment before symptoms appear, when research suggests it may be most effective. For more information on what to do about a positive SMA screen, visit our Support & Care section.
Prenatal Testing
Prenatal testing is used to determine if a fetus has inherited a genetic disorder. After consultation with their doctor, some families may choose this type of testing if their child is known to be at risk for SMA. One of two different tests may be used:
- In amniocentesis, the most common form of prenatal testing, a very fine needle is inserted into the woman’s abdomen, and amniotic fluid is extracted. This fluid contains fetal DNA that can be tested for SMA. Amniocentesis can be performed after the 14th week of pregnancy, and is associated with a risk of miscarriage that may be as high as 1 in 200.
- Chorionic Villus Sampling can often be performed as early as the 10th week of pregnancy. Chorionic villi are small, finger-like structures that form the placenta. Chorionic villi contain fetal DNA that can be extracted and tested for SMA. CVS is associated with a risk of miscarriage that may be as high as 1 in 100.
Carriers of Spinal Muscular Atrophy
Most people have two functioning copies of the SMN1 gene. People with one faulty copy and one functioning copy are called “carriers.”
Carriers generally do not show signs and symptoms of SMA, but could be at risk to have a child affected with the condition.
Approximately 1 in 50 people is a genetic carrier for SMA. Most carriers have no idea they are carriers until they have a child born with SMA.
How is SMA Inherited?
SMA is an autosomal recessive genetic condition. This means that a child must inherit two non-working copies of the SMN1 gene, typically one from each parent, in order to have SMA.
When two parents are carriers, there is:
- A 25% chance that their child will be unaffected
- A 50% chance that their child will be a carrier
- A 25% chance that their child will have SMA
If only one parent is a carrier, the child is usually not at risk for SMA, though they do have a 50% risk of being a carrier. However, in very rare cases, spontaneous genetic changes in the SMN1 gene can occur during egg or sperm production. In this situation, only one parent will be a carrier. In addition, a small percentage of carriers have genetic changes that cannot be identified through current testing technology. In this case, it will appear as though the disease has been caused by a single carrier.
Carrier Testing
A DNA test is the only way to know if a person is a carrier. The DNA test is a simple procedure, based on a blood test. In the general population, this test can detect about 95% of carriers. However, in African-American populations, detection is closer to 70%. This is because a difficult to detect mutation is seen more frequently in African-American populations than in other races.
The American College of Obstetricians and Gynecologists recommends that all women who are thinking about becoming pregnant or who are already pregnant be offered carrier screening for SMA and other genetic conditions. In addition, individuals with a family history of SMA are encouraged to have carrier screening. Deciding whether or not to undergo genetic testing is highly personal, and we strongly recommend discussing it with a physician or genetic counselor. Carrier screening via saliva testing is also available as an alternative to a blood test.
Reproductive Choices
For couples who are carriers, reproductive decisions can be sensitive. A number of options are available, such as prenatal testing, adoption, and pre-implantation genetic diagnosis (PGD). PGD screens embryos for genetic disorders and selects the unaffected embryos for implantation.
We believe that your family has the right to choose whatever option is best for you.
We help families understand their options and provide resources to support their decision-making process. We do not advocate any specific course of action, nor do we pressure families to choose one way or the other.
We encourage each family to discuss their situation with a an expert physician and a genetic counselor and only then take a final call.
TREATMENT AND CURE
Until 2016 spinal muscular atrophy was a disease to which there was no cure or any approved treatment, except for managing the symptoms and preventing complications. Spinraza is the first drug to be approved by the U.S. Food and Drug Administration (US FDA) on 23rd December 2016, to treat children and adults with spinal muscular atrophy.
Evrysdi (Risdiplam) by Roche pharmaceuticals, is the first and only treatment for adults and children two months or older suffering from SMA, now available in India. It is the only DCGI approved drug launched in India in July 2021 for SMA. Evrysdi was first approved by the US FDA in August 2020 and is today available in India within 11 months of the US approval. Evrysdi (Risdiplam) works by targeting the SMN2 gene (survival motor neuron gene 2), causing it to make more functional SMN protein. This increases SMN protein levels throughout the central nervous system and body, helping to improve motor nerve and muscle function in children and adults with spinal muscular atrophy (SMA). It is administered orally at home daily. Evrysdi must be taken for a person’s lifetime.
The price ranges from Rs 22Lakhs to 72Lakhs per year, depending upon the age and weight of the person.
Cure SMA India is working extensively for GST exemption on this drug.
Spinraza is given via an intrathecal (IT) injection, which is an injection into directly into the cerebrospinal fluid through the lower back. Individuals receive four “loading doses” within the first 2 months of treatment. Once those loading doses are completed, they receive a maintenance dose every 4 months for the duration of the individual’s life.
The drug is designed to increase production of the full-length SMN protein, which is critical for the maintenance of motor neurons.
The current price for Spinraza is $750,000 (Rs 5crores) for the first year and $375,000 (Rs 3 crores) for every year after for the life of the patient. It is approved for all forms of SMA.
This drug is unavailable in India.
Zolgensma is the only approved gene therapy for spinal muscular atrophy. It consists of an artificial SMN1 gene that is loaded into a specially prepared virus. The virus is injected in the veins or the spinal cavity where it “infects” the target cells with a functional copy of the SMN1 gene.
The therapy has been developed by a US company AveXis (now a subsidiary of Novartis). It has been approved for use in the United States on 24th May 2019 in children with SMA under 2 years. A single dose of AVXS-101 contains trillions of specially crafted viral particles that carry a synthetic SMN1 sequence (a transgene) inside a typical viral shell (capsid). Upon administration, these modified viruses “infect” neuronal cells with the SMN1 transgene. The transgene then starts working within the cell to produce fair amounts of SMN protein exactly the way a natural SMN1 gene would. In this way, AVXS-101 addresses the root cause of spinal muscular atrophy, that is the deficiency of SMN protein.
Novartis, set the price at $2.1 million (Rs 15crores), the most expensive drug ever.
A few patients in India are getting zolgensma under the GMAP (Global Managed Access Program).